液晶のMD計算
目次
はじめに
岸川先生と共同研究で行った棒状分子のMD計算を行うためのメモです。MD計 算までの手順としては、以下のとおりです。
- LigParGen使って棒状分子にOPLS-AA力場を割り当てたlammps dataを生成する。
- z軸を長軸となるよう分子を回転させてセル内に配置し、lammps inputs を生成する。
- lammpsでMD計算を実行する
1は自前で開発したものもありますが、力場パラメータも適時更新されてい るようなので、Web上のLigParGenをありがたく使用させてもらいます。 LigParGenは、mol/pdbファイルしか受け付けてくれないので、G16等で作成し た構造ファイルは、Open Babelを使って、molファイルに変換します。2は、 力場を割り当てた分子をセルに配置します。3はlammpsを別途インストール しておく必要があります。また以下のスクリプトは大窪が作成したものです。
作成したプログラムは、gitで配布してます。
git clone https://amorphous.tf.chiba-u.jp/gitweb/lab03/LC.git
molファイルの作成
分子描画ツールで1分子のmolファイルを作成する。g16やxyzファイルをmol ファイルに変換したい場合、Open Babelを使ってmolに変換できます。
obabel -ixyz ole-B-B-B-ole.xyz -omol > a.mol
VESTA等で、molのbond情報を確認すると良い。
lammpsdataの生成
molファイル(6-BB-6.mol)をLigParGenにsubmitして力場の割 り当てられたlmpdataを生成してもらって、ダウンロード(UNK_BC3019.lmp)する。 1.14*CM1A-LBCCの方が精度が良さそうですが、原著論文を読んで中身を理解 する必要があります。
格子の設定とlammpsファイルの生成
lmp2lmpin.pyに力場を割り当てたlmpdataを渡す。lmp2lmpin.pyは、次の処 理を行う。
- OPLS-AAの割り当てた部分電荷の総和が微妙に0にならないので、余った電 荷は全原子に割り当てて調整する。
- 直方体の占有体積が最小でz方向が長軸になるように分子を回転させる。
- 格子定数と格子点を指定して、単位格子を作る。格子定数と格子点(Pまた はC)は、引数で指定できる。
- 格子定数a, b, cは、引数で指定しなければ、分子サイズ(lx, ly, lz)か ら、a=lx, b=ly, c=lz+1.0とする。
- 作成した格子に基づいてlammps用インプットファイルを生成する。なお、 lammps用の.inと.dataを生成する。.inの中身はスクリプト中で定義して いる。
力場を割り当てたUNK_BC3019.lmpをlmp2lmpin.pyに渡して実行する。-oで出 力ファイルのbasenameを指定する。
lmp2lmpin.py UNK_BC3019.lmp -o test
これで、UNK_BC3019.lmpから次のファイルが生成される。test.cifは構造の 確認用に生成している。分子が近すぎず離れすぎず、結晶に近い初期構造に なっているかtest.cifを見て確認する。
- test.in
- test.data
- test.cif
なおスクリプトを実行すると標準出力に次のような構造情報が出力される。 格子定数と格子点を設定するために、分子の占有体積(サイズ)や、指定し た格子での密度を参考にすると良い。格子定数と格子点(PまたはC)は、引数で渡せる ので、cifファイルを確認しながら微調整する。
----------------------------------------------------------------------------
total charge (OPLS): 0.000300
total charge (correction): 0.000000
Molecule size: [ 5.08643041 3.50255321 32.70876729]
Molecule volume: 582.7228192674309
----------------------------------------------------------------------------
test.cif was created.
test.data was created.
----------------------------------------------------------------------------
Cell info:
a, b, c: (5.086430405850203 3.5025532085938877 33.70876729281664)
alpha, beta, gamma: (90 90 90)
Layer space: 33.70876729281664
Lattice points:
(0.0000 0.0000 0.0000)
Density: 1.2571482374902023 g/cm3
Replicate: [3, 3, 1]
Molecular info:
Atoms: 75
Bonds: 76
Angles: 138
Dihedrals: 192
Impropers: 45
Molecules: 1
Total charge: 0.000000
----------------------------------------------------------------------------
test.in was created.
(1/2, 1/2, 0)に格子点(C)を作って、(a, b, c)=(7, 7, 70)の格子にする場合は、引数で指定する。
lmp2lmpin.py UNK_BC3019.lmp -o test -a 7 -b 7 -c 60 --lattice_type C
germanium上でlammps実行
各ノードで計算させるために、job schedularを使用して計算jobを投げます (queingする)。以下のjob制御コマンドを使用して、queueされたjobを制御 する。qsubはフィルタースクリプトsubmit_lammps.py経由で呼び出すように しているが、直接コマンドで投入するこもと可能。
- qsub (jobの投入)
- qstat (jobの状態確認)
- qdel (jobの削除)
job投入フィルタースクリプト
作成したlammpsのinputからjobを投入しやすいようにsubmit_lammps.pyを 作成した。lammps inputを作成したら、次のコマンドでjobを投入できる。
submit_lammps.py test.in
これで、次のように表示される。
submit shell script: test.sh was created ---------------------------------------------------------------------------- #!/bin/sh #PBS -V #PBS -l nodes=1:ppn=4 #PBS -N test #PBS -j oe #PBS -k oed #PBS -q silicon #PBS -m n cd $PBS_O_WORKDIR OMP_NUM_THREADS=1 NPROCS=`wc -l < $PBS_NODEFILE` lammps=/usr/local/bin/lmp_silicon mpirun -np $NPROCS $lammps -in test.in -log test.log ---------------------------------------------------------------------------- A job script is OK? [y/n]
yを押せばjobが投入される。jobが投入されたらqstatで状態を確認する。jobのお知らせメールを受け取りたい場合は、-eでe-mailアドレスを指定する。また、node数を増やしたい場合は-nで指定する。
submit_lammps.py test.in -e ohkubo.takahiro@faculty.chiba-u.jp -n 2
jobが実行されると、job IDのついたファイルが生成される。途中の計算結果を参照した場合は、このファイルをlogplot.py等で確認する。
計算結果の確認
dump2cif.py
.lammpstrjはVMDで可視化できる。計算結果は、test.lammpstrjとtest.log に生成される。dump2cif.pyを使ってtest.lammpstrjの最終ステップからcif ファイルを作成する。これで、laststep.cifが生成される。
dump2cif.py test.lammpstrj -o laststep.cif
atomicdata.py
atomicdata.pyを使って生成したcifから分子をバラしたxyzファイルを作る。
atomicdata.py laststep.cif
これで、分子毎のxyzファイル(16個)が生成される。
- 0000.xyz
- 0001.xyz
- 0002.xyz
- 0003.xyz
- 0004.xyz
- 0005.xyz
- 0006.xyz
- 0007.xyz
- 0008.xyz
- 0009.xyz
- 0009.xyz
- 0010.xyz
- 0011.xyz
- 0012.xyz
- 0013.xyz
- 0014.xyz
- 0015.xyz
logplot.py
logファイルの統計情報を確認した場合は、logplot.pyにlogファイルを渡す。 これで最終ステップの統計情報が出力される。
logplot.py test.log
logのデータをexcel(-x)とpng(-p)に出力したいなら、引数で指定する。
logplot.py test.log -x -p
各runでの系情報をsheetで分けたtest.xlsxが生成され、3番目のrunで得ら れたデータがプロットされる。以下はd001のプロット例を示す。
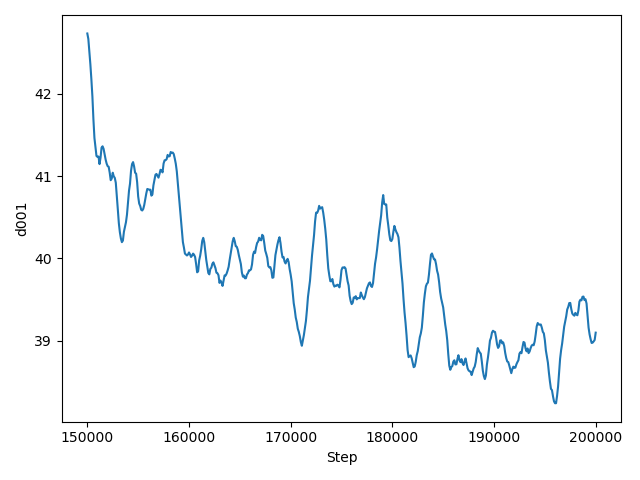
trj2vec.py
分子の部位を貫くベクトルを生成する。分子の部位は input_vector_info.xlsxを作成して、test.lammpstrjと一緒に引数で渡す。
trj2vec.py test.lammpstrj -i input_vector_info.xlsx
これで、VMDでベクトル可視化用のdraw.tclと各ステップでのベクトルをま とめたtest_vec.xlsxが生成される。生成したベクトルデータから、order parameterや分子の屈曲性、折りたたみ等を評価すれば良いでしょう。
ベクトル可視化用にdraw.tclもされます。test.lammpstrjの最終ステップ のベクトルデータを可視化します。VMDでtest.lammpstrjをloadしたら、 Extensions -> Tk consoleとすすんで、ターミナルを開いたら生成した draw.tclを読み込む。
source draw.tcl
これで指定した部位のベクトルが可視化される。
order_p.py
trj2vec.pyで生成したtest_vec.xlsxとlogから、order parameterや分子の dihedralを計算する。-lでlogファイルを渡す。温度、d001、エネルギー、エンタ ルピーもlogから拾ってデータをまとめる。
order_p.py -l test.log 300K-400Kc1_vec.xlsx
これでresult.xlsxが生成される。各部位についてのorder parameterは、ス テップ毎にmeanとstdがシートを分けて保存される。
dump2dihedral.py
lammpstrjから4つの原子タイプを指定して、cis-とtrans-を考慮した2面角を 計算する。4つの原子は結合している必要はない。モノマーに割り当てられた 原子タイプは、下図を参照する。
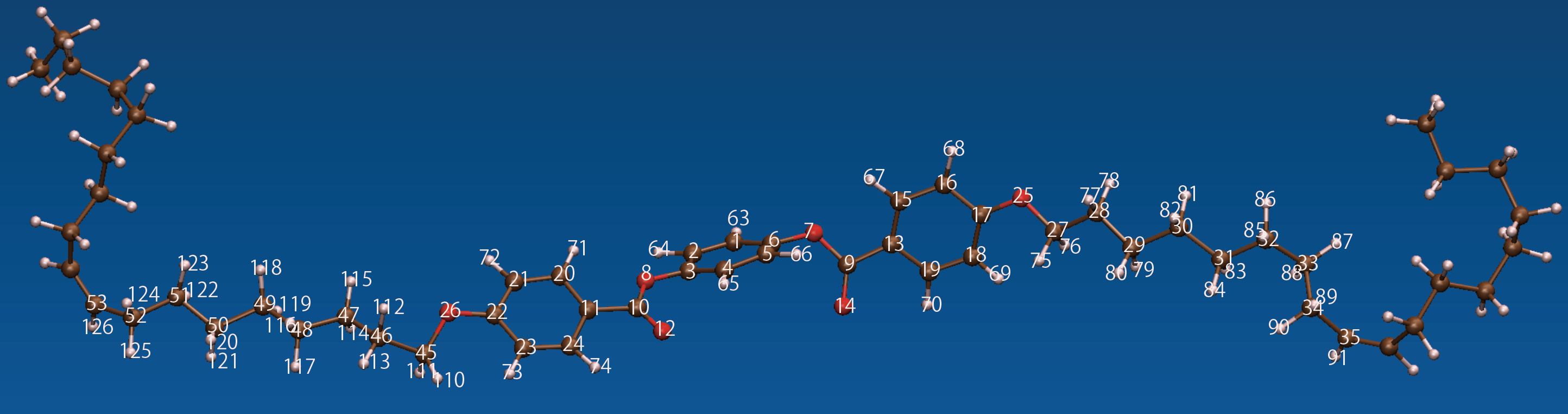
図1: モノマーの原子タイプ
dihedralのcisとtransは、dihedral\(\phi\) が次の条件になる。
- cis: \(|\phi| < 90\)
- trans: \(|\phi| > 90\)
dihedralの+と-方向は、lammpsの定義と反対方向としている。つまり、1-2-3 番目原子が作る平面を考えて、2と3番目原子のzは、z2<z3とし、1番原子のxは x軸と平行とする。このとき、4番目原子のxyからdihedralを計算している。
計算したいdihedralの4原子タイプは、ラベルに続いて4つのタイプ番号を並べ る。4原子タイプの指定は、複数セット指定できる。例えば、次の2つdihedral を計算したいとする。
- ラベルはleftで、原子タイプ 3-8-10-12
- ラベルはouterで、原子タイプ 12-3-6-14
この場合、次のように指定する。
dump2dihedral.py OLE-BBB-OLE-10101ir24-300-5ns.lammpstrj -d left 3 8 10 12 inner 12 3 6 14
これで次のようなdihedralの統計情報が出力される。
Total steps: 501
Total molecules: 100
Target types for dihedral calculation
left: [ 3 8 10 12]
inner: [12 3 6 14]
isomer C isomer C/% isomer T isomer T/% dihedral ave dihedral std
left 50100.0 100.000000 0.0 0.000000 0.707792 13.723843
inner 18666.0 37.257485 31434.0 62.742515 0.558539 113.277483
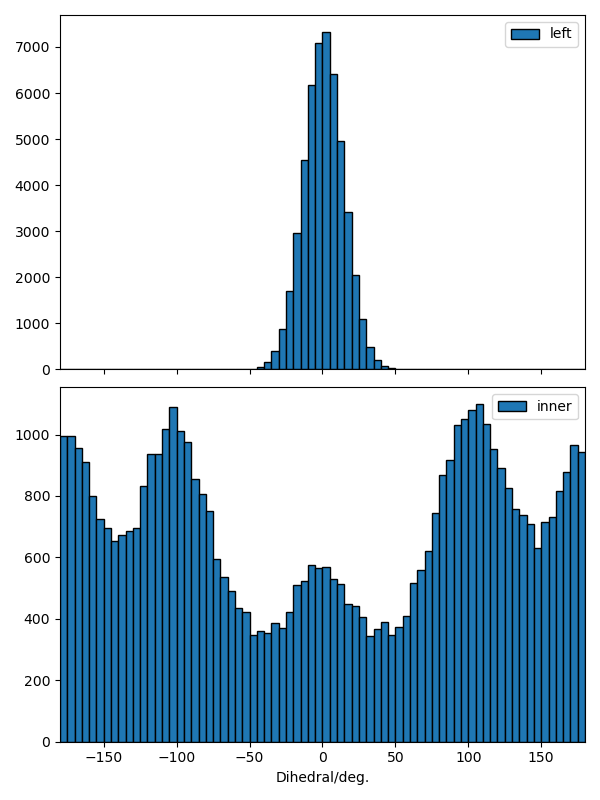
計算結果のすべてをExcel sheetにまとめて出力した場合は-x、dihedralのヒストグラ ムをプロットした場合は、-oで指定する。例えば、データdihedral.xlsxに出力 して、図をdihedral.pngに保存したい場合は、
dump2dihedral.py OLE-BBB-OLE-10101ir24-300-5ns.lammpstrj -d left 3 8 10 12 inner 12 3 6 14 -x dihedral.xlsx -o dihedral.png
これですべてのdihedralのデータを指定したラベルでまとめたdihedral.xlsxと dihedral.pngが出力される。dihedral.pngのデータは、dihedral.xlsxの histogramシートに保存されている。
{kind=link}
ヒストグラムのbin幅のdefaultは5 deg.にしている。10 deg.に変更したければ、-bで指定する。
./dump2dihedral.py OLE-BBB-OLE-10101ir24-300-5ns.lammpstrj -d left 3 8 10 12 inner 12 3 6 14 -x dihedral.xlsx -o dihedral.png -b 10
計算例
OLE-B-B-B-OLE
- OLE-B-B-B-OLE.mol molファイル
- UNK_86CB74.lmp LigParGenで生成したlammps input
分子をx軸回り45°回転させて、格子定数を指定し、3x3x2のスーパーセルのインプットファイルを生成する。
lmp2lmpin.py UNK_86CB74.lmp -a 5 -b 8 -c 35 --replicate 3 3 2 --molrotate 45 0 0 --cell_type tri -o test